作者丨福建医科大学附属协和医院 邹漳钰
来源丨神经科的那些事(ID:hsneuro)
病例介绍
女性,48 岁,进行性双下肢无力 7 年。初始症状为行走时左下肢无力感,1 年后出现左小腿萎缩伴肉跳感,右下肢行走无力伴右小腿肌肉萎缩;此后 6 年双下肢无力逐渐加重,肉跳感范围扩大。既往史无特殊,父母身体健康,否认父母近亲婚配史,否认有遗传病家族史。
体格检查:神清,言语清晰,对答切题,定向力、记忆力和计算力正常,无舌肌萎缩及纡颤,舌肌顶颊肌力 5,其余颅神经未见阳性体征,颈软,双上肢肌力 5 级,双下肢近端肌力 4 级,远端背屈肌力 4 级,跖屈肌力 4-级;双上肢肌张力正常,双下肢肌张力减低,双下肢肌肉萎缩,双上肢腱反射对称++,双下肢膝反射偏活跃,四肢深浅感觉检查正常,双侧 Hoffman 征阳性,右侧掌颌反射阳性,双侧 Babinski 征、chaddock 征阳性。
定位诊断思路的初步形成:见图 1
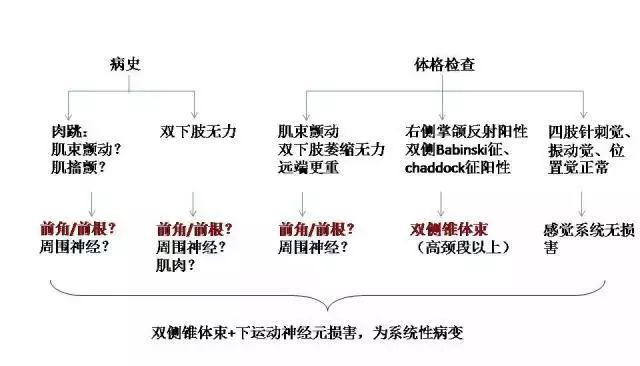
定性诊断思路的形成、需动用的处理检查:见图 2
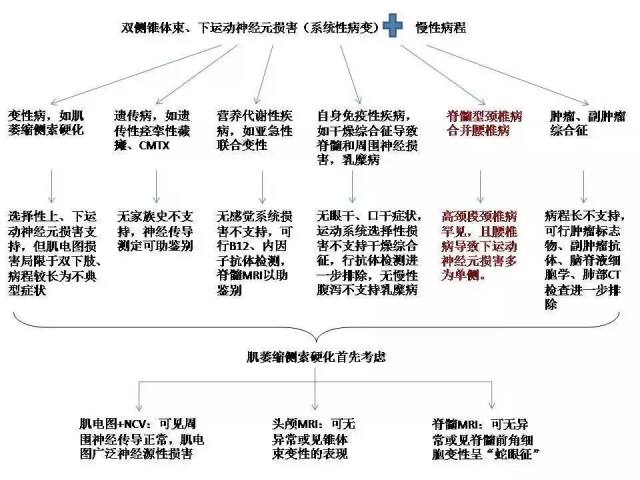
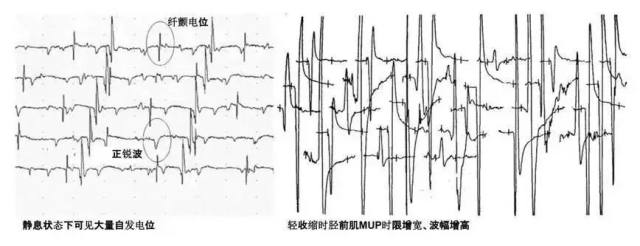
病人神经传导测定提示上、下肢周围神经运动和感觉传导均正常,针极肌电图示双下肢神经源性损害(双侧胫前肌、股内侧肌可见大量自发电位及高波幅、宽时限的 MUP,图 3)。
进一步行血生化、甲状腺功能、自身免疫抗体、肿瘤标志物、副肿瘤抗体均正常,脑脊液常规、生化、细胞学正常,双下肢 SEP 正常,肺部 CT、腹部彩超等检查未见明显异常,颅脑、颈椎、腰椎 MRI 未见明显异常。
详细的辅助检查排除其他鉴别诊断后,仍考虑 ALS 的诊断,鉴于患者病情发展缓慢,病程长达 7 年,下运动神经元损害仍局限于双下肢,症状不典型,行 ALS 相关基因检测,结果发现 SOD1 基因已知的致病突变 p.H46R 突变。
鉴于该类遗传性 ALS 为常染色体显性遗传而患者没有家族史,考虑为新发突变。
最终诊断:肌萎缩侧索硬化(SOD1 基因 p.H46R 突变)
基于本例的问题:
1. 临床上表现为「肉跳」的病理情况有哪些?
临床上表现为「肉跳」的病理情况包括肌束震颤(fasciculation)、肌颤搐(myokymia)和肌涟渏(ripping),但这三种病理情况的发生机制和表现截然不同。
肌束震颤是肌肉静息时由单个运动单位的不规则自发放电引起的,为轴突支配的一组肌纤维的不规则收缩。肌束震颤可以有不同的范围和幅度,较明显的肌束震颤肉眼即可看到,小的肌束震颤病人能感觉到但肉眼观察不到。
肌电图可以在静息状态下见到束颤电位,可伴随正锐波或纤颤电位,神经超声也可以发现肉眼难以发现的肌束颤动。病理性肌束震颤通常伴随肌肉无力和萎缩,常见于前角细胞病变、神经根病或周围神经病。
肌颤搐(myokymia)是一个或几个运动单位的重复放电引起的肌纤维自发性抽搐,皮肤下可见缓慢持续不规则的肌肉波纹状起伏或蠕动,肌纤维收缩沿肌纤维纵轴方向呈波浪样前进。
肌电图显示同一运动单位自发和重复放电,频率 2-60 次/秒,持续数秒钟。肌颤搐常见于慢性神经损伤轴突末端,常见于放射性臂丛神经病、Guillain-Barre 综合征等脱髓鞘周围神经病、中毒性周围神经病、脊髓病,和 Issacs 综合征等。
肌涟渏可能是由于骨骼肌通道机械敏感或牵张激活,叩击或牵张诱发的异常肌肉收缩。
表现为运动诱发的肌肉僵硬、间歇性疼痛和痉挛,肌肉在叩击或收缩后伸展时可诱发持续性「滚动」收缩,犹如水面涟漪一般扩展,通常肢体近端肌肉如股四头肌和肱二头肌最为明显。
肌肉涟漪般收缩的速度 0.6m/秒,并且涟漪收缩的方向与肌纤维长轴呈直角,可持续 5-20 秒,直接叩击肌肉尚可诱发叩击性肌强直,也常伴腓肠肌肥大,肌电图检查静自状态下插入电位轻度增加,而涟漪样收缩时呈电静息。
常见于涟漪性肌病(包括肢带型肌营养不良 1C 的亚型以及重症肌无力相关获得性等)。
2、ALS 的临床表型包括哪些?
ALS 并非单一的疾病,而是包括了一组异质性疾病的综合征,其临床表型主要包括仅累及下运动神经元的进行性肌萎缩(PMA),仅累及上运动神经元的原发性侧索硬化(PLS),同时累及上、下运动神经元的经典型 ALS,局限于延髓的进行性延髓麻痹(PBP),局限于上肢的连枷臂综合征(FAS),局限于下肢的连枷腿综合征(FLS),局限于一侧肢体的偏瘫型 ALS,以及局限于四肢远端的假性多神经炎型 ALS。
3、ALS 如何诊断?
ALS 目前还没有特异性的诊断生物学标志物,因而诊断主要是临床诊断。临床上用得比较多的诊断标准是在 1998 年修订的 El Escorial 诊断标准基础上提出的 Awaji 诊断标准,诊断标准如下表:

ALS 的诊断标准中强调随诊及鉴别诊断的重要性,对于处于疾病早期或症状不典型的病例随诊发现病变向其他节段进展可帮助临床确诊。
随着近年来越来越多的 ALS 相关致病基因的发现,2015 年再修订的 El Escorial 诊断标准中明确提出,如果基因检测发现已知的 ALS 相关基因的致病突变,可在仅存在一个区域上运动神经元或下运动神经元损害的情况下诊断 ALS。
这充分说明了随着新技术的出现,ALS 的诊断标准从临床、电生理维度向分子生物学维度拓展。
4、ALS 的鉴别诊断包括哪些疾病?
ALS 的鉴别诊断主要根据患者的临床表现,同时存在上、下运动神经元损害的患者需要和以下疾病鉴别:颈椎病、副肿瘤综合征和乳糜病等。
以上运动神经元损害为主要临床表现的患者需要和以下疾病鉴别:遗传性痉挛性截瘫(HSP)、亚急性联合变性(SCD)和肾上腺脑白质营养不良等。
以下运动神经元损害为主要临床表现的患者需要和以下疾病鉴别:平山病(HD)、脊髓性肌萎缩症(SMA)、脊髓延髓肌萎缩症(SBMA)或肯尼迪病(KD)、脊髓灰质炎后综合征(PPS)、多灶性运动神经病(MMN)、Lewis-Summer 综合征、己糖胺酶 A 缺乏症、包涵体肌炎(IBM)和面肩肱型肌营养不良(FSHD)等。
5、哪些疑诊 ALS 的患者需要进行基因检测?
并非所有疑诊 ALS 的患者都需要进行基因检测,下面几种情况可考虑进行基因检测:
单纯下运动神经元损害需要排除 SMA 和 KD 时;
有明确家族史的患者;
存在特殊临床表型的患者,如病情进展特别快、生存期特别短,或者病情进展特别慢、生存期特别长,或者合并痴呆/额颞叶痴呆。
本例病人发病 7 年后肢体无力仍局限于双下肢,肌电图也仅见局限于双下肢的神经源性损害,生活尚可自理,病程较良性,症状不典型,因此行基因检测得以确诊。
需要指出的是目前已发现的基因突变约占家族性 ALS 的 2/3,散发性 ALS 的 10%,因此,即使基因检测未发现致病突变也不能藉此排除 ALS 的诊断。
6、SOD1 基因致病的主要机制是什么?
SOD1 基因是首个被发现的 ALS 的致病基因,编码的 SOD1 蛋白属于超氧化物歧化酶家族,主要功能是抗氧化,阻止自由基损伤细胞。
SOD1 突变的致病机制仍尚不明确,最初认为 SOD1 突变引起 ALS 的可能机制是突变的 SOD1 酶活性减低,导致自由基清除障碍,继发氧化损伤运动神经元,但后来的研究发现 SOD1 突变主要通过「功能的获得」导致 ALS,突变的 SOD1 蛋白异常折叠、聚集形成聚合物,沉积于运动神经元的细胞质中,导致细胞氧化应激损伤增加、兴奋性毒性增加,启动或促发神经元调亡。
7、ALS 目前主要的治疗方法是什么?
目前 ALS 的治疗主要强调的是综合治疗。药物治疗方面,利鲁唑作为目前唯一的病因治疗药物,通过阻滞突触后膜的谷氨酸受体和 NMDA 受体,可以延缓疾病的发展。
研究显示该药可使 ALS 患者的生存期延长 3-6 个月,另外,根据患者的症状个体化给予对症治疗药物,可以缓解痉挛、疼痛、焦虑、大量唾液分泌导致的流涎等症状。
护理也是治疗的一个重要组成部分,早期无创呼吸机的使用可以改善病人的通气不足;对吞咽困难或出现饮水呛咳的病人早期行经皮胃造瘘(PEG)可以保证充足的能量充分供应,避免由于能量不足消耗肌肉,预防吸入性肺炎,改善病人的生活质量,提高生存期。
8、ALS 的预后如何?
ALS 患者平均生存期为 3-5 年,但个体差异较明显,约 10% 患者生存期可达 10 年以上。流行病学研究显示发病年龄小、临床表现以上运动神经元损害为主的患者预后相对较好,而老年起病、低体重指数、延髓起病的患者预后较差。
ALS 患者的预后也与其临床表型密切相关,PLS 预后最好(生存期多超过 20 年),FAS、FLS 和 PMA 的预后也较好(多数生存期 5 年以上),而延髓起病的 ALS 预后最差(生存期 2-3 年)。
ALS 患者的生存期还与其携带的基因突变有关,如 SOD1 基因 p.D90A 和 p.H46R 突变患者生存期相对较长(甚至可长达 30 多年),而 SOD1 基因 p.A4V 突变和 FUS 基因 p.P525L 突变、移码突变患者生存期较短(绝大多数小于 2 年,最短 6 个月)。此外,利鲁唑和无创呼吸机治疗也可使 ALS 患者的生存期轻度延长。
参考文献
1.de Carvalho M, Dengler R, Eisen A, England JD, KajiR, Kimura J, et al. Electrodiagnostic criteria for diagnosis of ALS.ClinNeurophysiology. 2008;119:497-503.
2.Ludolph A, Drory V, Hardiman O, Nakano I, Ravits J,Robberecht W, et al. A revision of the El Escorial criteria-2015.Amyotrophic lateral sclerosis and frontotemporal degeneration. 2015;16:291-2.
3.Swinnen B, Robberecht W. The phenotypic variability of amyotrophic lateral sclerosis. Nature reviews Neurology. 2014;10:661-70.
4.ChenL, ZhangB,ChenR,TangL,LiuR,YangY, et al. Natural history and clinical features of sporadicamyotrophic lateral sclerosis in China.Journal of Neurology NeurosurgeryPsychiatry.2015;86:1075-81.
5.Zou ZY, Cui LY, Sun Q, Li XG, Liu MS, Xu Y, et al.De novo FUS gene mutations are associated with juvenile-onset sporadic amyotrophic lateral sclerosis in China. Neurobiology of Aging.2013;34:1312.e1-8.
6.Zou ZY, Liu MS, Li XG, Cui LY. H46R SOD1 mutation is consistently associated with a relatively benign form of Amyotrophic Lateral Sclerosis with slow progression. Amyotrophic lateral sclerosis and frontotemporal degeneration. 2016. In press.
编辑|俊华
文章转载授权及合作事宜请联系微信「panda_wqy」